1
ContigScape User Manual 1.0
Introduction ................................................................................................................................................................. 2
Highlights ..................................................................................................................................................................... 2
Installation ................................................................................................................................................................... 3
Overview....................................................................................................................................................................... 3
Open files ...................................................................................................................................................................... 4
Open CRS file........................................................................................................................................................ 4
Open 454Contig.ace file ........................................................................................................................................ 5
Open AGP file ....................................................................................................................................................... 7
Open mate-paired reads ......................................................................................................................................... 7
Alignment Options: ........................................................................................................................................... 8
Quality threshold: ............................................................................................................................................ 8
Minimum read length: ..................................................................................................................................... 8
Minimum quality score to keep: ...................................................................................................................... 8
Minimum percent of bases: ............................................................................................................................. 8
Number of CPU for BWA: .............................................................................................................................. 8
Visualization Options .................................................................................................................................................. 9
Show contig width by ratio .................................................................................................................................... 9
Show contig sequences: ......................................................................................................................................... 9
Hide contig link < {value} .................................................................................................................................... 9
Hide contig < {value} bp....................................................................................................................................... 9
Repeat contig threshold ......................................................................................................................................... 9
Single contig threshold .......................................................................................................................................... 9
Visualization appearance .......................................................................................................................................... 11
Repeat contig color for >2x: ................................................................................................................................ 11
Probable contig color for >1.5x: .......................................................................................................................... 11
Single contig color for <1.5x: .............................................................................................................................. 12
Contig shape for <2kb: ........................................................................................................................................ 12
Link label size: .................................................................................................................................................... 12
Link color: ........................................................................................................................................................... 12
Link label color: .................................................................................................................................................. 12
Link color for gap: ............................................................................................................................................... 12
Link label for gap: ............................................................................................................................................... 12
Set shape for plasmid:.......................................................................................................................................... 12
Maximum link width: .......................................................................................................................................... 12
Minimum link width: ........................................................................................................................................... 12
Show/Hide result ........................................................................................................................................................ 13
Show intermediate results .................................................................................................................................... 13
Estimate Plasmid/TIRs ........................................................................................................................................ 14
Contig sequences........................................................................................................................................................ 15
Algorithm principle ................................................................................................................................................... 16
Finding connections between 454 reads .............................................................................................................. 16
Finding connections between mate-paired reads ................................................................................................. 16
Genomic features prediction ................................................................................................................................ 17
Copyright and Contact.............................................................................................................................................. 18
Frequently Asked Questions (FAQs) ........................................................................................................................ 19
1
Introduction
Thank you for choosing ContigScape, which is a Cytoscape plugin that offers construction and visualization of de novo
genome assemblies. Its intuitive manner makes it very suitable for discovering contig relationships, and therefore helpful in
gap-closing strategy. The program is freely available from http://sourceforge.net/projects/contigscape/files/.
Highlights
Automatically construct possible connections between scaffolds or contigs using assembly of reads (ACE format),
mate-paired reads, scaffolding information (AGP format) or user defined CRS files. ContigScape lets you manipulate the
appearance of the graph, as well as filter the contigs by their length and coverage. Meanwhile, the sequence contigs of
plasmids and special repeats (IS elements, ribosomal RNAs, terminal repeats etc) can be displayed as well. The following
figure illustrates network appearance upon loading ACE file, mate-paired reads and AGP file, respectively.
2
Installation
Before using ContigScape, you need to make sure you have Java and Cytoscape2.8.3 installed. After that, you just need to
put ContigScape.jar under installation directory cytoscape/plugins folder or browse to plugin using “Install plugin from
file” which is under Plugins menu.
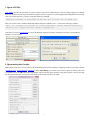
Overview
ContigScape is loaded automatically along with Cytoscape, which can be found in the left panel. The graphic interface is
divided into four categories: “Open files”, “Visualization Options”, “Visualization appearance”, “Show/Hide result”,
“Contig sequences”. There are several options under each category,. You can click the categories to make options hide or
show up.
I
II
III
V
IV
3
I Open files allows user to choose different file types to import into ContigScape. Currently ACE format, mate-paired
reads, AGP format, and user defined tab-delimited files are supported.
II Visualization Options allows user to check which type of graphic object you wish to show on the graph, as well as see
statistical information of the graph.
In able to obtain the best visual effects, please use “Layout”—“Force-Directed
Layout”
button
and “Visual style” —“sample1”
III Visualization appearance allows user to control the appearance of the graph, such as color and size for different types
of nodes/edges.
IV Show/Hide result allows user to export intermediate result and estimate plasmid/TIRs.
V Contig sequences allows user to check selected contig sequences. It is also possible for users to send the sequences to
NCBI Blast upon selection.
I. Open files:
1 Open CRS file:
CRS format includes two files, and each contains three columns. ’tabbed.txt’ represents the number of connections among
contigs, and ‘tabbedCov.txt’ describes all the contigs’ length and coverage. User can input ’tabbed.txt’ guiding how
contigs are connected to each other. This is usefully in creating connections using diverse data from other excellent software,
or adds additional connections to the existed network. The following example shows how to prepare ’tabbed.txt’ files
using the beginning and end of the contig to create connections. Please notice do not include connections between the
beginning and end of the contigs unless they are self-connected (the contig maybe a plasmid, etc), as contig140 in the
following example. The format of ’tabbed.txt’ is as follows:
Begin
3S
3E
140S
End
119E
58E
140E
Connections-ReadsNum (optional)
113
322
29
NOTICE: “S” represents “start position”, “E” represents “end position”
‘tabbedCov.txt’ file can be imported as additional information by checking “input tab-delimited contig coverage file” and
specify the location to the file. The file should also be tab-delimited and follow the format in the table below.
Contig ID Length Coverage (or copy number)
119
169
40.72
(1.94)
3
3295
24.73
(1.18)
58
31775 21.92
(1.04)
4
The CRS test files of 10 454Contigs.ace can be downloaded from
http://sourceforge.net/projects/contigscape/files/datasets/454Contigs_CRS_test_files.zip
2 Open 454Contig.ace file:
ACE files are generated by various assembly programs, including Phrap, CAP3, Newbler, Arachne, AMOS, etc. ACE file
not only includes contigs and reads of the assemblies, but also contains information of the connections between the contigs.
Take 454Contig.ace for example, which was generated using 454 reads and Newbler program. Upon pressing “Open
454Contig.ace”, a dialog promotes the user to input the locations to the sequence file. The graph below indicates the
connections/copy number of the contigs after import 454Contig.ace.
5
6
3 Open AGP file:
AGP format describes the assembly of a larger sequence object from smaller objects, such as joining contigs into scaffolds.
Here it is useful in forming larger assemblies using the reference sequences. You can apply BLAST alignment of the contigs
onto close-related reference sequences using the following command.
blastall -p blastn -i contig.fas -d ref.fas -m 8 -e 1e-30 –F F -o output.txt
Then you need to remove both the duplicated and the ambiguous matches (score < 1000 in the following example):
perl -ne '@t=split(/\t/);next if $t[11]<1000;print unless defined $h{$t[0]};
$h{$t[0]}=1;' output.txt|sort -k9,9n > output2.txt
After that you can use blast2agp.pl to convert the BLAST output into AGP file, and then press button “open AGP file”
and browse to its location:
4 Open mate-paired reads:
Mate-paired reads can be used to construct the relationship between the scaffolds. ContigScape needs several tools to do this
(SAMtools, bwa, FASTX-Toolkit, BEDTools). In the Open Dialog, user needs to specify the sequences and the path to the
executables. Press the OK button and ContigScape will precede the input sequences using the programs. After processing,
the files generated will be saved in the same folder as the contig sequences you input.
7
Alignment Options
The options below are for setting the parameters used while mapping mate-paired reads onto the contigs. For the program
used, please also refer to http://hannonlab.cshl.edu/fastx_toolkit/commandline.html#fastx_trimmer_usage and
http://bio-bwa.sourceforge.net/bwa.shtml.
Quality threshold:
Set quality to be trimmed (from the end of the sequence) by fastq_quality_trimmer
Minimum read length:
Sequences shorter than this will be discarded by fastq_quality_trimmer.
Minimum quality score to keep:
Minimum quality score to keep by fastq_quality_filter.
Minimum percent of bases:
Minimum percent of bases filtered by fastq_quality_filter.
Number of CPU for BWA:
Number of threads used by bwa while doing alignment, see also “bwa aln”.
8
II. Visualization Options:
Show contig width by ratio:
Check this option to apply modification to contigs width according to their length. Otherwise contigs are shown in the same
width.
Show contig sequences:
Store contig sequences in memory while loading 454Contigs.ace files. The contig sequences are shown in the data panel
below on click the nodes/edges representing each contig.
Hide contig link < {value}
Hide the connection for those contig connections having a reads count lower the value specified by user. Check this option
if user want to leave high confident connections only.
Repeat contig threshold
Define repeat threshold, default value is 2
Single contig threshold
Define specific contig, default value is 1.5
9
10
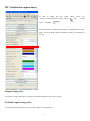
III. Visualization appearances:
In able to obtain the best visual
“Layout”—“Force-Directed Layout” button
effects, please use
and
“Visual
style” —“sample1”
The options below control the appearance of the graph, such as color,
shape, size of the graph objects and labels according to constrains set
by user.
Repeat contig color:
Set color for contigs which have coverage greater than two-folds of the overall coverage.
Probable repeat contig color:
Set color for the contigs whose coverage is greater than 1.5 and less than 2.
11
Single contig color for:
Set color for contigs which have coverage lower than 1.5-folds of the overall coverage.
Contig shape for <2kb:
Set shape for contigs which have length greater than 2k base pairs.
Link label size:
Set size for the labels of the contig connections.
Link color:
Set color for contig connections.
Link label color:
Set color for the labels of contig connections.
Link color for gap:
Set color for connections of gaps.
Link label for gap:
Set color for labels of connections of gaps.
Set shape for plasmid:
Set shape for the contigs predicted as plasmid.
Maximum link width:
Set maximum width for the connections between contigs. Set to 25 by default.
Minimum link width:
Set minimum width for the connections between contigs. Set to 0.12 by default.
12
IV. Show/Hide result:
Show intermediate results:
Intermediate results contain information generated while loading ACE files, AGP files and mate-paired reads. The following
example shows contig and contig connections information for ACE file loaded. User can choose either “Export” to save
current table, or “Export All” to save all the tables.
13
Estimate Plasmid/TIRs:
Plasmid results include predicted plasmids with coverage and type information.
14
V. Contig sequences
Show sequence in the data panel field below upon clicking corresponding contigs and edges.
15
When viewing the graph, the 1,000 base pairs of both 5’-end and 3’-end can be loaded, with 20 “N” linking them
representing the middle sequences. Clicking the edge of two contigs, the sequence containing corresponding contigs’ ends
can also be displayed. The displayed sequence can be used to design primers in ContigScape and perform blast against
NCBI database.
Algorithm principle
Finding connections between 454 reads:
To our knowledge, Roche 454 shotgun sequencing is the most suitable strategy for de novo genome sequencing. The reads
length of Roche 454 reached 700bp now and could resolve small repeats caused gaps. But reads of longer repeats would be
assembled into only one contig and thus produced gaps in other repeat regions. The‘Newbler Assembler’ could produce a
‘454Contigs.ace’ file, which contained all the assembly information and could be opened by ‘Consed’ (Gordon, D. et
al. 1998). As we can see from the following, when a read was separated into two contigs, the coordinate of the read in each
contig will be shown after the read name, followed by the contig number this read linked, using‘fmX’representing 5’ end
of the read located in contigX and ‘toY’ representing 3’end of the read located in contigY. This label showed a unique
advantage of‘Newbler Assembler’ endowed by 454 long reads. We can extract all the ‘fm’ and ‘to’information from
‘454Contigs.ace’ file, and arrange them into a relationship table, such as ‘5’-end-Contig1’ linked ‘3’-end -Contig2’.
Then the table could be displayed by ContigScape.
Finding connections between mate-paired reads:
material:matePairedReads1.fq, matePairedReads2.fq, matePairedContigs.fas, scaffold.pl
The most common way to assemble contigs into scaffolds is through mate-pair information. Depending on the fragment size
16
of mate-pair reads, scaffolding program can identify how far two contigs should be apart from each other. For example, if
two contigs were separately mapped by a pair of 3kb apart mate-pair reads, the two contigs could be joined into a scaffold
and the gap size should be 3kb minus distance from mapping loci to the end of contigs. The repeat region less than 3kb
could be spanned using this method. But if the repeat region was longer than the fragment size of mate-pair library,
ambiguous linkage information would occur. Same as ‘Newbler’ result of 454 reads, the ContigScape could display a
network relationship within scaffolds by counting mate-pair reads number linking LargeContigs (>500bp). Detailed method
was described in scaffold.pl (http://sourceforge.net/projects/contigscape/files/datasets/).
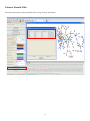
Genomic features prediction:
Genomic features can be categorized into several groups: plasmids, phage elements, and terminal repeats, which can be
accessed through “show Plasmid/TIRs” in the control panel. See the following figure for details. In Figure A. The interface
of ContigScape. The left part was the control panel, the window on the right shows a sample genome. Contigs were
indicated by red (repeated contig) , dark blue (unique contig) and orange( probable repeats). B. Zoomed image of some
contigs (light blue frame in panel A). B1. A linear plasmid formed by three contigs. B2. Repeats (Contig28) in the end of
chromosome. B3. A circular plasmid with high copy number formed by three contigs (Contig141,142,143) . B4. Two
high-copy number circular plasmids each formed by a single contig. B5. A linear plasmid with high copy number formed by
one contig. B6. A circular plasmid with single copy number composed of one contig.
Judging whether a repeat contig was from chromosome or plasmid mainly depended on the linkage information of two
ends of this contig. Four different types were shown in Figure, 1). Repeat contigs connected in a circular fashion (Panel 3),
2).Individual contig connected itself without anyone else (Panel 4 and 6), 3). One end of repeat contig having no linkage to
any other contigs, usually representing linear chromosome telomere or linear plasmid end (Panel 1 and 2), 4).A linear
plasmid composed of only one repeat contig without connections to any contigs (Panel 5). While if a plasmid is linear and
single copy, ContigScape cannot distinguish it. We can estimate whether or not a contig was a plasmid effectively based on
above described situation in our experience. Of course researcher must confirm whether it is a plasmid or not by PCR,
sequencing and annotation.
In Figure B, 143E has connections with 142E and 144E (Panel 3). But the number of connections (800) between 143E
and 142E is more than that (10) between 143E and 144E. In this case, the latter might be a nonspecific connection caused
by little overlap among the reads. Additionally, Figure 6B shows that contig78 in the linear plasmid
80E-80S-78E-78S-54E-54S also has another copy in the chromosome (Panel 1).
17
Copyright
ContigScape is a free software; you can redistribute it and/or modify it under the terms of the Lesser GNU General Public
License as published by the Free Software Foundation; either version 2 of the License, or (at your option) any later version.
This program is distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY; without even the implied
warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the Lesser GNU General Public
License for more details.
You should have received a copy of the Lesser GNU General Public License along with this program; if not, write to the
Free Software Foundation, Inc., 59 Temple Place, Suite 330, Boston, MA 02111-1307 USA
Contact
You may email any questions/suggestions to: [email protected], [email protected]
You're welcome to report bugs at ContigScape Discussion Forums.
18
Frequently Asked Questions(FAQs)
1. Why the network looks messed up after I opened 454Contigs.ace?
A: You have to do layout manually after loading the network, one way is to use "Force directed layout" in Cytoscape
menu. In able to obtain the best visual effects, please use “Layout”—“Force-Directed Layout”
button
and
VizMapper-“Visual style” —“sample1”
2. How do I make an AGP file by comparing the contigs to the reference sequences?
A: You may do a local BLAST between the contig and the reference sequences with “-m 8” parameter, then use a
script called "blast2agp.pl" included in this software package to convert the BLAST output to an AGP file.
3. Have GRASS, SSPACE and OPERA been integrated into the package of ContigScape?
A: Sorry, we have not integrated them. User can prepare scaffold result handled with these tools to CRS format file
mentioned in the above, and then ContigScape will provide a Global visualization.
4. I had some problem to install the java class. It doesn’t work on my Mac, and gives me an error. On a Linux system I got
it to run. I loaded the 454 contigs as ace file into ContigScape. Further, it is extremely “slow”. My java job is always on
99%..
A: It looks that you have not install java correctly. Please download Java 1.6 or higher version from
http://java.sun.com and install it properly.
5. You mentioned contigs can be reordered, broken and the results can be saved. I haven’t found those functionalities in
ContigScape. Can the results be saved?
A: Those functions can be found in Editor panel of Cytoscape. Cytoscape is a software package with perfect
visualization network and high operability. That is why we select it.
6. Would it be possible to import bam files? Those contain most of the relevant information, and it is a widely used format.
A: You can use bamToBed in BEDTools package to convert a sorted bam file into a bed file, then specify both the
contig sequence and the bed file from "open mate-paired reads" dialog to load.
7. I saw the scaffold.pl script, but why can’t it be run within ContigScape?
A: Scaffold.pl is a perl script used in processing ‘fastq’ file and corresponding contig file with the purpose of gaining
a scaffold.bed and tabbed.txt including contigs’ relationships. Scaffold.pl needs Fastx_toolkit, BEDTools, BWA,
Samtools which were developed only based on Linux platforms. In fact, user can run scaffold.pl in “open
mate-paired reads ” of ContigScape on Linux platforms.
8. In the abstract you said that you can find repeats like IS elements, Ribosomal RNA etc. This is detection purely done by
coverage of reads? If due to long reads like PacBio, those are assembled into their single copies, will you still detect them?
A: It’s true. The contig number of bacterial genome will greatly reduced using PacBio technology. ContigScape
won't be able to detect small repeat like IS elements, Ribosomal RNA. But, large repeat contig still exist in several
bacterial genome, fungi genome and all plant and animal genome. Large repeats more than 10kb usually have
important biological significance.
9. The scope is supposed to be microbial genomes gap closing, but would the tools be able to be applied to scope with plant
and animal genome with ten thousands of contigs?
A: User may convert the result to CRS format file mentioned in the above, and then ContigScape provides a Global
visualization. Though thousands of contigs may look complicated in the image, it is the real relationship among these
contigs.
19
10. How do I get "454Contigs.ace" for my assembly?
A: You have to ensure you checked "Single ACE file" or "Single ACE file for small genomes" in GS De novo
Assembler Output Sub-Tab, see
http://454.com/my454/documentation/gs-flx-system/emanuals/Part_C/wwhelp/wwhimpl/common/html/wwhelp.htm#
href=PartC.1.016.html&single=true
11. Why I cannot obtain the result after input scaffold.bed and corresponding contig sequence ?
A: Please check the integrity of the scaffold.bed and make sure that the sequence title in scaffold.bed is same as
corresponding contig sequence.
12. Can I install it in Cytoscape 3.0?
A: Sorry, ContigScape 1.0 don’t support Cytoscape 3.0 now, we will update it in version 1.1.
13. I have not clearly understood whether the program was specific to Newbler assembler or not.
A: The “open 454Contigs.ace” option can be only used to input 454Contigs.ace, which had been modified in
ContigScape v1.0. Ace format is a very large file with various contents. We cannot process so many formats
sand we provided CRS format described in the manuscript to fit different program. CRS format contains
two
files,
and
users
can
refer
to
http://sourceforge.net/projects/contigscape/files/datasets/454Contigs_CRS_test_files.zip/download.
14. How it provides help in finishing after viewing a graph?
A: Contigs displayed in ContigScape were better operational, as the repeat contigs, gaps and even plasmids can be
highlighted, filtered, and customized. So, graph itself will facilitate a faster and more precise determination of the
linkages among contigs and greatly improve the efficiency of gap closing.
After view a graph, we can get the sequence of contigs’ ends which may be used to blast and designing primer.
Click the edge of two contigs, the sequence containing corresponding contigs’ ends can be used to blast on NCBI to
judge the edge is true or not. Other, if user needs to edit the connections of the network, the user can open “edit
panel” to edit. All the gaps needs to be filled by PCR and sequencing using ABI3730 sequencer. We have added this
point in part “Display functionality of ContigScape” of our revision.
In our finishing strategy, all contigs together with ABI3730 data must be assembled using phred, phrap, consed at
last. Our plugin cannot replace “consed” program. It looks like a canvas which used to edit and judge the order
among contigs and can evaluate the complexity of assembly visually.
20